
From GB to ISFG: allele notation correction for LongTR and HipSTR
LongTR and HipSTR report STR alleles using base-pair differences from the reference, not standard forensic allele numbers. This article explains the math behind the discrepancy, shows why allele 11.8 is actually allele 11.3 in forensic databases, and provides the conversion rule for tetranucleotide, trinucleotide, and pentanucleotide loci.
When LongTR or HipSTR is used for STR genotyping, the allele numbers in the output sometimes differ from those in reference databases, CE-based profiles, or other forensic tools. A locus that should return allele 11.3 at D2S441 may appear as 11.8. No error is raised. The software has performed correctly. The discrepancy is a consequence of two incompatible naming conventions applied to the same underlying sequence, and it can be resolved with a straightforward arithmetic conversion.
What is the GB field?
LongTR and HipSTR do not store allele numbers directly in the VCF. Instead, they record the number of base pairs by which each allele differs from the reference sequence at that locus. This value is stored in a field called GB, short for GenotypingBases.
A real vcf output line looks like this:
4|1:-1|8:1.00:0.50:16:0:0:0|0:0|0:28.03:-2|1;-1|5;8|8;9|1;12|1:-1|6;8|10
The GB field is the second segment: -1|8
The pipe symbol separates the two alleles:
1means allele 1 is 1 base pair shorter than the reference+8means allele 2 is 8 base pairs longer than the reference
Every allele number in the final genotype is derived from these two values together with the reference information in the BED file used during genotyping.

For D2S441 in hg38, the relevant BED file values are:
ref_repeats = 12(the reference carries 12 complete repeat units)period = 4 bp(the motif is TCTA, four bases long)- Reference size = 12 × 4 = 48 bp
Walking through the calculation
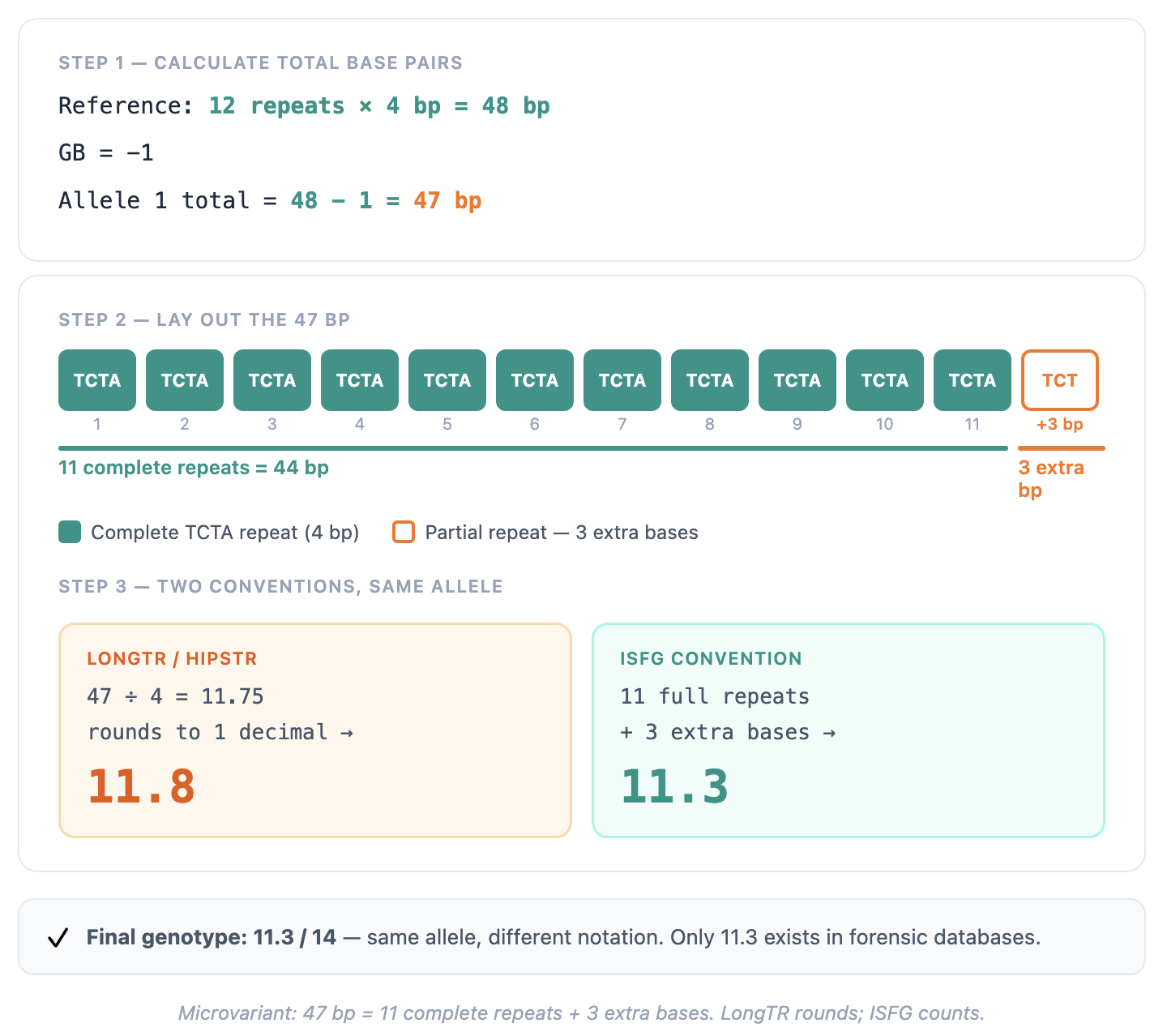
Following allele 1 (GB = -1) step by step:
Step 1. Total base pairs of this allele:
Reference: 12 repeats × 4 bp = 48 bp GB = -1 Allele 1 total = 48 - 1 = 47 bp
Step 2. This is where LongTR and ISFG diverge.
LongTR divides total base pairs by the motif length and rounds to one decimal place:
47 ÷ 4 = 11.75 → LongTR reports: 11.8
ISFG convention asks a different question: how many complete repeat units fit into 47 bp, and how many bases remain?
47 bp = 11 complete TCTA repeats (44 bp) + 3 extra bases ISFG notation: 11.3
Both values describe the same physical sequence of 47 base pairs. The difference lies entirely in how the partial component is expressed: LongTR reports a rounded decimal fraction of a repeat unit, while ISFG reports the count of extra bases directly.
Allele 2 requires no correction: GB = +8 → 48 + 8 = 56 bp → 56 ÷ 4 = 14.0 → no partial repeat → ISFG: 14.
Why this matters
Forensic population databases, including CODIS reference data, ESS frequency tables, and published population studies, use ISFG notation exclusively. An allele designated 11.8 does not appear in any of these resources. Submitting LongTR output without prior conversion will return no database match and produce no error message. The discrepancy is silent and, if undetected, will affect likelihood ratio calculations and cross-platform profile comparisons.
The same issue arises when validating long-read results against CE-based profiles from the same individual or against known reference samples.
The conversion rule
The relationship between LongTR decimals and ISFG notation is fixed and depends only on the motif length. For tetranucleotide loci, which represent the majority of the standard forensic panel:
| LongTR decimal | Extra bases | ISFG suffix |
|---|---|---|
| .3 | 1 | .1 |
| .5 | 2 | .2 |
| .8 | 3 | .3 |
The pattern holds because LongTR computes extra bases ÷ period and rounds. For a tetranucleotide locus, 3 extra bases gives 3÷4 = 0.75, which rounds to .8. ISFG writes the same situation as .3, counting the extra bases directly. The full conversion table for trinucleotide and pentanucleotide loci is available in the STRhub Tools section.
Confirmed affected loci when using the standard HipSTR hg38 BED file:
LongTR .8 → ISFG .3: D1S1656, D2S441, D6S1043, D12S391, TH01
LongTR .5 → ISFG .2: D18S51, D19S433, D21S11, FGA
Summary
LongTR and HipSTR encode allele information as a base-pair difference from the reference sequence, stored in the GB field of the VCF. The allele size is obtained by applying this difference to the reference size and dividing by the motif length. The resulting decimal is rounded to one place by the tool. ISFG notation bypasses the division and instead records the number of extra bases that remain after counting complete repeat units. For tetranucleotide loci, a LongTR value of .8 invariably represents 3 extra bases, which ISFG designates as .3. This conversion must be applied before any database query, likelihood ratio calculation, or cross-platform profile comparison.
References
- Frontanilla Recalde T, Ayala Jacquet JM, Mendes Junior CT et al. (2026). HipSTR-UI: a web-based interface for forensic STR genotyping with long-read sequencing data. Forensic Science International: Genetics. https://doi.org/10.1016/j.fsigen.2026.103456
- Valle-Silva G, Frontanilla T, Ayala J, Donadi EA et al. (2022). Analysis and comparison of the STR genotypes called with HipSTR, STRait Razor and toaSTR by using next generation sequencing data in a Brazilian population sample. Forensic Science International: Genetics, 58, 102676. https://doi.org/10.1016/j.fsigen.2022.102676
- Gettings KB, Bodner M, Borsuk LA, King JL, Ballard D, Parson W et al. (2024). Recommendations of the DNA Commission of the International Society for Forensic Genetics (ISFG) on short tandem repeat sequence nomenclature. Forensic Science International: Genetics, 68, 102946. https://doi.org/10.1016/j.fsigen.2023.102946
- Willems T, Zielinski D, Yuan J, Gordon A, Gymrek M, Erlich Y. (2017). Genome-wide profiling of heritable and de novo STR variations. Nature Methods, 14(6), 590–592. https://doi.org/10.1038/nmeth.4267 Ziaei Jam H, Zook JM, Javadzadeh S et al. (2024). LongTR: genome-wide profiling of genetic variation at tandem repeats from long reads. Genome Biology, 25, 176. https://doi.org/10.1186/s13059-024-03319-2