
From Length to Sequence: CE vs NGS in STR Analysis
CE defines STR alleles by size, while NGS reveals their true sequence. This added resolution improves discrimination and mixture interpretation.
STR alleles are traditionally defined by fragment length using capillary electrophoresis (CE). This approach has been the foundation of forensic DNA analysis for decades. Understanding what changes when moving from length-based to sequence-based STR typing is essential for interpreting modern forensic data.
The length-based model (CE)
In CE-based STR typing, alleles are assigned based on the size of amplified DNA fragments. Fragment length reflects the number of repeat units present, and allele numbers are inferred accordingly.
CE measures fragment length, not sequence.
This model is robust, standardized, and highly reproducible across laboratories. For routine forensic casework, it remains the operational standard.
The resolution limit of length
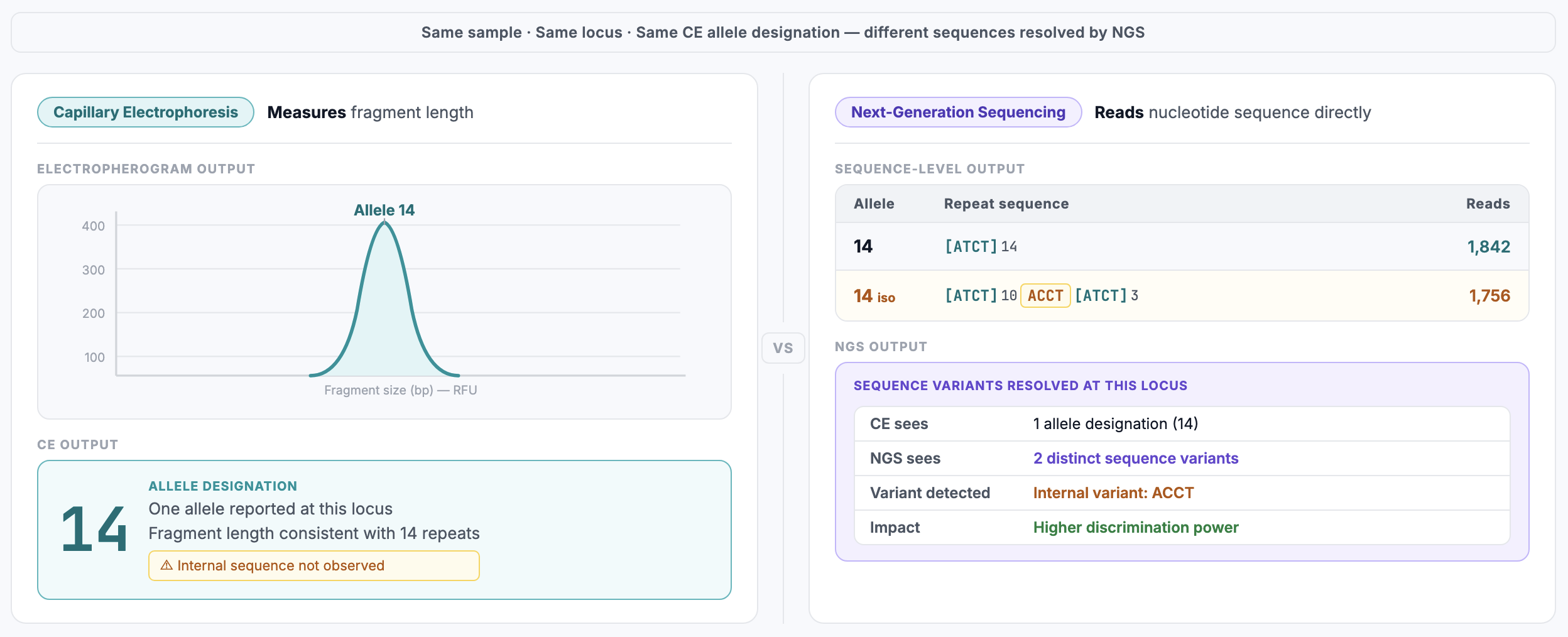
Different DNA sequences can produce fragments of identical length. These are known as isoalleles; alleles that share the same CE designation but differ at the sequence level.
Example:
[ATCT]₁₄ — 14 perfect repeats
[ATCT]₁₂[ATCG]₁[ATCT]₁ — internal sequence variation
Both are reported as Allele 14 by CE. This is not a limitation of instrument sensitivity, it is a structural constraint of size-based typing. Same length does not imply identical sequence.
The sequence-based model (NGS/MPS)
Next-generation sequencing (NGS), also referred to as massively parallel sequencing (MPS), directly reads the nucleotide sequence of each STR allele. Instead of inferring allele size, NGS captures the repeat structure, any internal sequence variation, and the flanking sequence context.
NGS measures sequence, not just length.
As a result, alleles that appear identical in CE can be distinguished at the sequence level.
What sequence resolution adds
Moving from length to sequence has four concrete consequences for forensic STR analysis:
Isoalleles become distinguishable. Alleles collapsed into a single CE designation are resolved into distinct sequence variants. Each variant is counted and reported independently.
Discrimination power increases. A greater number of distinct alleles at a locus means a lower probability of a random match. Published studies consistently report higher effective allele counts when sequence data replaces size data at commonly used forensic loci.
Mixture interpretation improves. Contributors who share the same CE allele may carry different repeat sequences. NGS resolves them as distinct alleles, providing additional information for mixture deconvolution that CE cannot offer.
Microvariants are fully characterized. CE detects microvariants by size, it can identify that allele 9.2 exists, but not which bases form the partial repeat. NGS provides the complete sequence, enabling precise and reproducible characterization.
When sequence-level resolution matters
For many routine single-source cases, CE provides sufficient resolution. Sequence-level information becomes critical in specific contexts:
- Complex DNA mixtures with two or more contributors
- Cases where discrimination power must be maximized
- Population studies requiring allele diversity at the sequence level
- Research contexts where isoallele frequency affects statistical models
Why CE remains the standard
CE remains the dominant technology in operational forensic laboratories for well-established reasons: it is fast, cost-effective, supported by decades of population databases, and governed by internationally harmonized interpretation guidelines. The transition to NGS does not make CE obsolete; it defines the context in which each technology is appropriate.
Barriers to NGS adoption
Despite its analytical advantages, NGS introduces challenges that explain its limited adoption in routine casework:
Bioinformatics requirement. Raw sequencing reads must be quality-filtered, aligned to a reference genome, and processed by STR-specific software before any allele can be called. This requires infrastructure and expertise not yet standard in most forensic laboratories.
Validation burden. Introducing NGS under forensic accreditation standards requires extensive internal validation; sensitivity thresholds, stutter characterization at the sequence level, mixture interpretation guidelines, and concordance with existing CE-based databases.
Absence of universal standards. CE benefits from decades of harmonized allelic ladders and inter-laboratory proficiency schemes. Equivalent infrastructure for sequence-level STR alleles is still under development.
Cost and throughput. For high-volume routine casework, CE remains faster and less expensive per sample.
NGS adds resolution and, also complexity. Both need to be weighed against the specific analytical question being addressed.
Direct comparison
| Criterion | CE | NGS / MPS |
|---|---|---|
| Output | Fragment length | Nucleotide sequence |
| Isoallele resolution | No | Yes |
| Microvariant characterization | Size only | Full sequence |
| Mixture resolution | Size-based | Sequence-level |
| Bioinformatics required | Minimal | Substantial |
| Throughput (routine) | High | Lower (batch) |
| Standardization | Established | Developing |
| Population databases | Extensive | Limited, growing |
Summary
- CE and NGS are not competing technologies, they operate at different levels of resolution and serve different analytical needs.
- When fragment length provides sufficient information, CE is the appropriate tool. When sequence-level resolution changes the outcome (complex mixtures, discrimination-critical cases, or population research) NGS provides information that CE structurally cannot
- The question is not which technology is better. The question is which level of resolution the case requires.