
Understanding Sequencing File Formats: An Introductory Guide
Understanding sequencing data storage formats in bioinformatics
For many researchers entering the field of bioinformatics and genomics, one of the first challenges is understanding the file formats. What is the difference between FASTA, FASTQ, SAM, BAM, and CRAM? What do they contain, and how do you work with them?
This confusion is normal at the beginning.
Sequencing technologies generate massive volumes of data, and different formats exist to store, organize, compress, and share this information efficiently. Learning what these formats represent is one of the first steps toward becoming fluent in bioinformatics.
In this article, we will provide a conceptual overview of the most common sequencing file formats. The goal is not to cover every technical detail, but to help beginners build a mental map of how sequencing data move through an analysis pipeline. Later articles will explore each format in depth.
The sequencing data pipeline
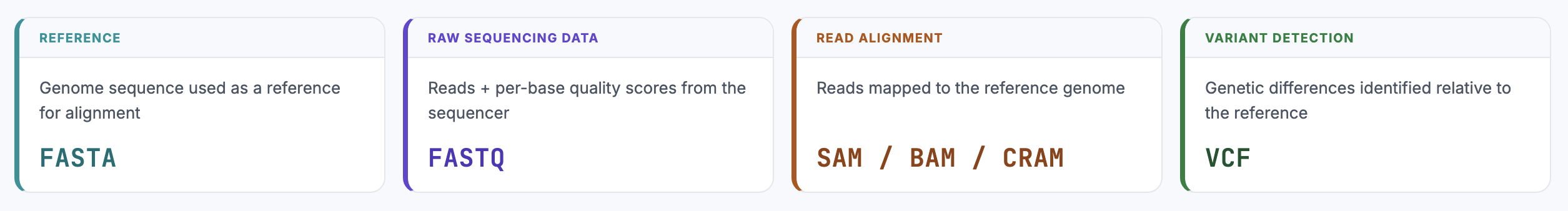
A simplified sequencing workflow looks like this:
- Reference genomes are stored in FASTA files
- Sequencing machines generate reads, which are stored in FASTQ files
- Reads are aligned to a reference genome, producing SAM, BAM, or CRAM files
- Genetic variants can then be identified from these alignments and stored in VCF files
Each format represents a different stage of interpretation of the same biological data.
The main sequencing file formats
FASTA
FASTA files store reference sequences such as genomes, genes, or contigs.
They contain only the sequence itself, without any quality information.
Example:
chr1
ATGCTTAGCTAGCTAGCTAGCTAGCTAG
Typical uses:
- Reference genomes
- Gene databases
- Assembled contigs
Think of FASTA as the map of the genome.
FASTQ
FASTQ files store raw sequencing reads together with quality scores for each base.
This is usually the starting point of most sequencing analyses.
Example structure:
@read_001
ACGTTCTGATGACCTTAGCA
+
IIHFGEFIIHDF>?=;:987
Each read has four lines:
| Line | Content |
|---|---|
| 1 | read identifier |
| 2 | nucleotide sequence |
| 3 | separator (+) |
| 4 | quality scores |
Quality scores estimate the probability of sequencing errors.
You can think of FASTQ as your raw material.
SAM
SAM (Sequence Alignment Map) files store how reads align to a reference genome.
They contain detailed information such as:
- Genomic coordinates
- Mapping quality
- Mismatches
- Insertions and deletions
Example (simplified):
read_001 0 chr1 10583 60 50M * 0 0 ACGTTCTGATGACCTTAGCA *
SAM files are human-readable text, but they can become extremely large.
BAM
BAM is simply the binary, compressed version of SAM.
It stores the same information but:
- Takes much less disk space
- Can be processed faster by software
Because of this, BAM is the standard working format in most genomic pipelines.
CRAM
CRAM is an even more efficient compressed format.
Instead of storing full sequences, CRAM files store differences relative to a reference genome, allowing additional compression.
Advantages:
- Much smaller file size
- Better long-term storage
- CRAM: the long-term archive to keep everything safe and compact.
This makes CRAM ideal for archiving large sequencing datasets.
Comparing alignment formats
SAM, BAM, and CRAM all store aligned reads, but differ in how they encode them.
| Format | Type | Size | Use |
|---|---|---|---|
| SAM | Text | Very large | Debugging / inspection |
| BAM | Binary | Smaller | Standard analysis |
| CRAM | Reference-compressed | Smallest | Long-term storage |
VCF
VCF (Variant Call Format) files store genetic variants detected from sequencing data.
Instead of containing full sequencing reads, VCF files record differences relative to the reference genome, such as:
- Single nucleotide variants (SNVs)
- Insertions and deletions (indels)
- Structural variants
Example:
chr1 879317 . G A 50 PASS .
VCF files, therefore, represent a higher level of interpretation, summarizing how a sample differs from the reference genome.
Typical uses include:
- Variant discovery
- Population genetics
- Clinical genomics
- Forensic genomics
You can think of VCF as a catalog of genomic differences.
Why are my samples ending with “.gz”?
Many sequencing files you download will appear with names like:
sample1_R1.fastq.gz sample1_R2.fastq.gz
The “.gz” extension does not indicate a different file format. Instead, it means the file has been compressed using gzip, a standard compression tool in Unix/Linux systems.
Why compress?
- Reduce file size
- Speed up downloads and transfers
- Save storage space
Final thoughts
Understanding sequencing file formats is not just about technology.
It directly affects how we work with genomic data.
The format used can influence:
- Storage efficiency
- Interpretation of results
- Reproducibility of analyses
This becomes especially important when studying Short Tandem Repeats (STRs), where the way sequencing data are stored and processed can affect how repetitive regions are interpreted.
For beginners, sequencing formats may seem intimidating at first.
But once you understand the logic connecting them, navigating genomic datasets becomes much easier.
Continue learning
If you would like to explore these formats further, STRhub includes additional articles that examine each sequencing file format in detail.