
Archivos FASTQ
Los archivos FASTQ son la base del análisis de secuenciación de nueva generación. Comprender cómo interpretar estas puntuaciones y cuándo aplicar el recorte (trimming) es esencial para garantizar análisis posteriores precisos en genómica y bioinformática.
¿Qué es un FASTQ?
Un archivo FASTQ almacena lecturas de ADN/ARN y su calidad por base. Siempre existe al menos un archivo FASTQ. Si el experimento fue paired-end, habrá dos archivos (R1 y R2). Si fue single-end, habrá solo uno (R1).
Cada lectura ocupa 4 líneas:
- ID de la lectura
- Secuencia (A/C/G/T/N)
- Separador (+)
- Calidad: una cadena de caracteres ASCII, uno por base, que codifica el puntaje Phred (confianza del base-calling)
**Los archivos FASTQ casi siempre se entregan comprimidos como .fastq.gz.
EJEMPLO
@A00469:123:HGF2KDSXX:1:1101:10003:12345 1:N:0:ACGTAC
ACGTTCTGATGACCTTAGCA
+
IIHFGEFIIHDF>?=;:987
- Línea 1: Identificador (instrumento, corrida, coordenadas, etc.). El 1 antes de :N: indica R1 (si fuera 2, sería R2).
- Línea 2: Secuencia (20 bases en este ejemplo).
- Línea 3: Separador (+).
- Línea 4: Calidad por base, del mismo largo que la secuencia (20 caracteres).
¿Cómo interpretar la calidad por base?
Está relacionada con la probabilidad de error P de la siguiente manera:
El puntaje de calidad Phred Q se define como:
Q = -10 · log10(P) ⇒ P = 10-Q/10
- Q10 → tasa de error de 1/10 (90% de confianza)
- Q20 → 1/100 (99%)
- Q30 → 1/1000 (99.9%)
- Q40 → 1/10.000 (99.99%)
Ejemplo de interpretación paso a paso
ACGTTCTGATGACCTTAGCA
IIHFGEFIIHDF>?=;:987
| Pos | Base | Carácter | ASCII | Q (Phred) | P(error) aprox.) | Interpretación |
|---|---|---|---|---|---|---|
| 1 | A | I | 73 | 40 | 0.0001 (0.01%) | Extremadamente confiable |
| 2 | C | I | 73 | 40 | 0.0001 | Extremadamente confiable |
| 3 | G | H | 72 | 39 | 0.00013 | Muy confiable |
| 4 | T | F | 70 | 37 | 0.0002 | Muy confiable |
| 5 | T | G | 71 | 38 | 0.00016 | Muy confiable |
| 6 | C | E | 69 | 36 | 0.00025 | Muy confiable |
| 7 | T | F | 70 | 37 | 0.0002 | Muy confiable |
| 8 | G | I | 73 | 40 | 0.0001 | Extremadamente confiable |
| 9 | A | I | 73 | 40 | 0.0001 | Extremadamente confiable |
| 10 | T | H | 72 | 39 | 0.00013 | Muy confiable |
| 11 | G | D | 68 | 35 | 0.00032 | Confiable |
| 12 | A | F | 70 | 37 | 0.0002 | Muy confiable |
| 13 | C | > | 62 | 29 | 0.0013 (0.13%) | Aceptable |
| 14 | C | ? | 63 | 30 | 0.001 (0.1%) | Aceptable |
| 15 | T | = | 61 | 28 | 0.0016 (0.16%) | Moderada |
| 16 | T | ; | 59 | 26 | 0.0025 (0.25%) | Moderada |
| 17 | A | : | 58 | 25 | Moderada–baja | Moderada–baja |
| 18 | G | 9 | 57 | 24 | 0.004 (0.4%) | Baja–moderada |
| 19 | C | 8 | 56 | 23 | 0.005 (0.5%) | Baja |
| 20 | A | 7 | 55 | 22 | 0.0063 (0.63%) | Baja |
Interpretación global
- Inicio de la lectura (posiciones 1–10): puntajes muy altos (Q37–40), prácticamente sin errores.
- Región media (11–14): la calidad disminuye levemente (Q29–35), aún aceptable.
- Final de la lectura (15–20): la calidad baja considerablemente (Q22–28), con una tasa de error esperada de 0.1–0.6%: esta es la región donde los secuenciadores Illumina típicamente presentan problemas.
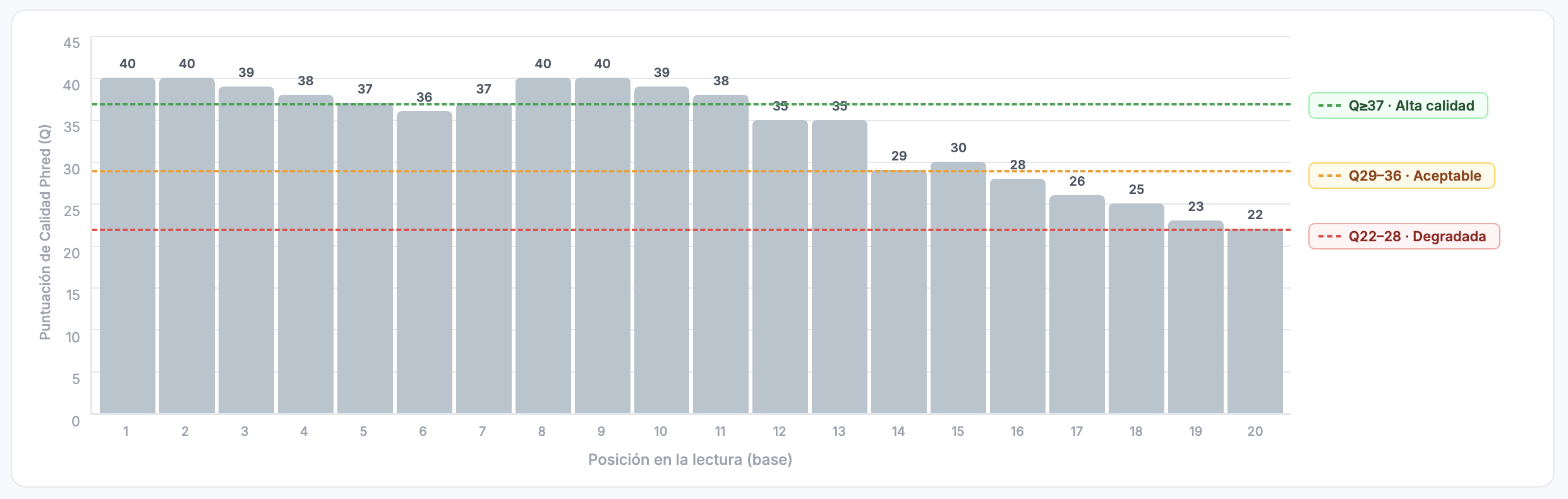
Este patrón es común: alta calidad al inicio, caída al final. Por eso a menudo se realiza trimming de los últimos nucleótidos.
EN RESUMEN
- FASTQ = datos brutos (bases + calidad por base).
- Siempre existe al menos un archivo FASTQ.
- Si el experimento fue paired-end, se secuenciaron ambas hebras, resultando en dos archivos (R1 y R2).
- Si fue single-end, habrá solo un archivo (R1).