
Arquivos FASTQ
Os arquivos FASTQ são a base da análise de sequenciamento de nova geração. Compreender como interpretar essas pontuações e quando aplicar o corte (trimming) é essencial para garantir análises subsequentes precisas em genômica e bioinformática.
O que é um FASTQ?
Um arquivo FASTQ armazena leituras de DNA/RNA e sua qualidade por base. Sempre existe pelo menos um arquivo FASTQ. Se o experimento foi paired-end, haverá dois arquivos (R1 e R2). Se foi single-end, haverá apenas um (R1).
Cada leitura ocupa 4 linhas:
- ID da leitura
- Sequência (A/C/G/T/N)
- Separador (+)
- Qualidade: uma string de caracteres ASCII, um por base, que codifica o score Phred (confiança do base-calling)
**Arquivos FASTQ quase sempre são entregues comprimidos como .fastq.gz.
EXEMPLO
@A00469:123:HGF2KDSXX:1:1101:10003:12345 1:N:0:ACGTAC
ACGTTCTGATGACCTTAGCA
+
IIHFGEFIIHDF>?=;:987
- Linha 1: Identificador (instrumento, corrida, coordenadas, etc.). O 1 antes de :N: indica R1 (se fosse 2, seria R2).
- Linha 2: Sequência (20 bases neste exemplo).
- Linha 3: Separador (+).
- Linha 4: Qualidade por base, com o mesmo comprimento da sequência (20 caracteres).
Como interpretar a qualidade por base?
Ela está relacionada à probabilidade de erro P da seguinte forma:
O score de qualidade Phred Q é definido como:
Q = -10 · log10(P) ⇒ P = 10-Q/10
- Q10 → taxa de erro de 1/10 (90% de confiança)
- Q20 → 1/100 (99%)
- Q30 → 1/1000 (99,9%)
- Q40 → 1/10.000 (99,99%)
Exemplo de interpretação passo a passo
ACGTTCTGATGACCTTAGCA
IIHFGEFIIHDF>?=;:987
| Pos | Base | Caractere | ASCII | Q (Phred) | P(erro) aprox. | Interpretação |
|---|---|---|---|---|---|---|
| 1 | A | I | 73 | 40 | 0.0001 (0.01%) | Extremamente confiável |
| 2 | C | I | 73 | 40 | 0.0001 | Extremamente confiável |
| 3 | G | H | 72 | 39 | 0.00013 | Muito confiável |
| 4 | T | F | 70 | 37 | 0.0002 | Muito confiável |
| 5 | T | G | 71 | 38 | 0.00016 | Muito confiável |
| 6 | C | E | 69 | 36 | 0.00025 | Muito confiável |
| 7 | T | F | 70 | 37 | 0.0002 | Muito confiável |
| 8 | G | I | 73 | 40 | 0.0001 | Extremamente confiável |
| 9 | A | I | 73 | 40 | 0.0001 | Extremamente confiável |
| 10 | T | H | 72 | 39 | 0.00013 | Muito confiável |
| 11 | G | D | 68 | 35 | 0.00032 | Confiável |
| 12 | A | F | 70 | 37 | 0.0002 | Muito confiável |
| 13 | C | > | 62 | 29 | 0.0013 (0.13%) | Aceitável |
| 14 | C | ? | 63 | 30 | 0.001 (0.1%) | Aceitável |
| 15 | T | = | 61 | 28 | 0.0016 (0.16%) | Moderada |
| 16 | T | ; | 59 | 26 | 0.0025 (0.25%) | Moderada |
| 17 | A | : | 58 | 25 | Moderada–baixa | Moderada–baixa |
| 18 | G | 9 | 57 | 24 | 0.004 (0.4%) | Baixa–moderada |
| 19 | C | 8 | 56 | 23 | 0.005 (0.5%) | Baixa |
| 20 | A | 7 | 55 | 22 | 0.0063 (0.63%) | Baixa |
Interpretação global
- Início da leitura (posições 1–10): scores muito altos (Q37–40), praticamente sem erros.
- Região intermediária (11–14): qualidade reduz um pouco (Q29–35), ainda aceitável.
- Final da leitura (15–20): queda considerável na qualidade (Q22–28), com taxa de erro esperada de 0,1–0,6% — exatamente onde sequenciadores Illumina costumam apresentar problemas.
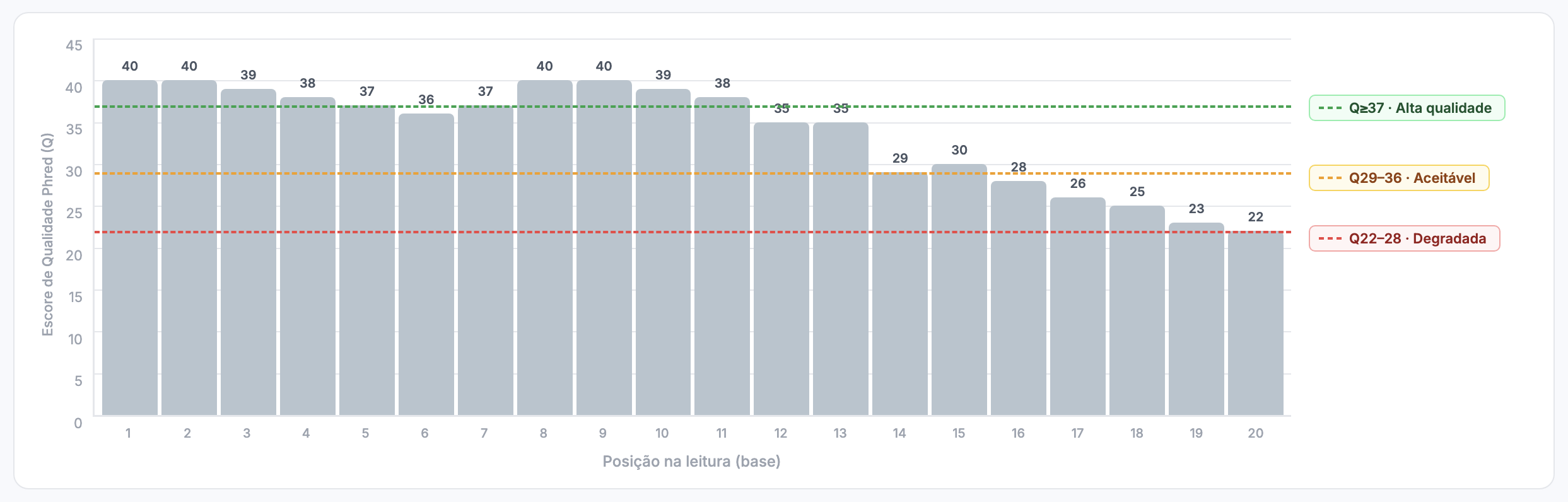
Esse padrão é comum: alta qualidade no início e queda no final. Por isso, o trimming dos últimos nucleotídeos é frequentemente realizado.
RESUMO
- FASTQ = dados brutos (bases + qualidade por base).
- Sempre existe pelo menos um arquivo FASTQ.
- Se o experimento foi paired-end, ambas as extremidades foram sequenciadas, resultando em dois arquivos (R1 e R2).
- Se foi single-end, haverá apenas um arquivo (R1).